Mitoxantrone: an agent with promises for anticancer therapies
Pritish Varadwaj, Krishna Misra, Anju Sharma, Rajnish Kumar
Department of Bioinformatics, Indian Institute of Information Technology Allahabad, Deoghat, Jhalwa, Allahabad-211012(U.P.), India.
Abstract
Mitoxantrone (MX) is an anthraquinone derivative with promising perspective in cancer chemotherapy. As compared to the better known anthraquinone derivatives of the anthracycline family, daunomycin and adriamycin, Mitoxantrone is comparatively more reactive, while being less cardiotoxic. Mitoxantrone recognizes the chromatin structure with higher affinity than free deoxy-ribonucleic acid (DNA) but mode of interaction is unknown. The intercalative binding of mitoxantrone in minor groove of DNA is much less favorable than in the major groove and also found to be much less sequence selective. Mitoxantrone shows considerable preference for intercalation of chromophore ring in a pyrimidine (3’- 5’) purine sequence rather than the isomeric purine (3’-5’) pyrimidine sequence. The insertion of mitoxantrone into DNA sequence is preferred in parallel mode. Formaldehyde plays an important role in the activation of mitoxantrone, leading to the formation of Drug-DNA adducts. These interstrand cross links take place in the presence of mitoxantrone and formaldehyde in a time and concentration dependent manner. It is specific to CpG and CpA sites in DNA, which prompts investigations into the effect of cytosine methylation (CpG) on adduct formation. The adducts at these methylated sites exhibited same stability as nonmethylated sites, suggesting that cytosine methylation simply increases drug accessibility to DNA rather than
Keywords
Mitoxantrone; anthraquinone; DNA; anticancer agent.
1. Introduction
Mitoxantrone (MX, Figure 1), a synthetic anticancer agent, is a member of the anthracenedione class of compounds and was originally designed as a simplified analogue of the anthraquinone-containing anthracylines [1,2]. The structure of MX is symmetrical which comprises of a tricyclic planar chromophore and two basic side chains [3]. The basic side groups of MX are critical for intercalation into DNA sequence and the quinine prosthetic group, which together could confer chemical reactivity. The positively charged N-containing side chains projects outwardly from the molecule and stabilizes the ring between base pairs by intercalating with the negatively charged phosphate backbone DNA [4]. MX structure is viewed as an anthracycline analog with the daunosamine sugar of adriamycin and other anthracycline being replaced with two identical amino alkyl side chains [1-3, 5-6]. It retains the aromatic ring structures of the anthracylines that permit intercalation into DNA. Binding of MX to DNA causes DNA condensation and inhibits DNA replication and RNA transcription. It also inhibits enzyme topoisomerase II, which is important for repair of damaged DNA resulting in the breakage of single and double strands [7].

Figure 1. Mitoxantrone, atom numbering (courtesy: K.X. Chen et al, 1986).
2. Mitoxantrone as an effective anti-cancer compound
MX shows an activity comparable to that of the better known anthraquinone derivatives of the anthracycline family, Daunomycin and adriamycin, while being less cardio toxic [3,8] and is effective against solid tumors, melanoma and non-small cell lung cancer [5]. MX also shows significant activity in patients with acute non-lymphocytic leukemia, advanced breast cancer, non-Hodgkins lymphoma [2]. MX offers comparable efficacy but lower acute toxicity than the most active currently available single agents used in treatment of advanced breast cancer [5].
Generally cardiotoxicity is associated with oxygen-derived free radicals that are generated by redox cycling of anthracycline quinine. It was observed that quinone further readily reduces to the semiquinone which reacts rapidly with oxygen and eventually leads to the production of hydroxyl and other oxygen radicals [6]. Heart tissues are sensitive to oxygen radical damage due to the absence of detoxifying enzymes such as catalase and superoxide dismutase [9]. In contrast to the above hypothesis associated with cardiotoxocity, Due to more negative reduction potential MX does not reduce to the semiquinone intermediate and therefore does not exhibit the cardiotoxicity, whereas anthracyclines, andriamycin and daunomycin exhibit cardiotoxicity. Molecular modeling studies on optimized structure of MX was carried out by Bhartwal et al [10] and showed that the torsional angles C7-C8-N11-C12 (φ1), C8-N11- C12-C13 (φ2) and N11-C12-C13-N14 (φ3) have very little flexibility. They are constrained to cis, trans and gauche+/gauche- conformations, respectively. They concluded that the gauche conformations of φ3 may be crucial for anticancer activity as various other MX analogues who readily adopt trans conformations are found to be less effective anti-cancer compounds than MX. The primary side effect associated with MX is nausea and vomiting and major delayed toxicity is myelosuppression [6,11].
2.1 Mode of interaction
The ultimate target of a drug when inserted into body either orally or intravenously, in a cell is DNA, with which drug interacts principally by intercalation causing DNA condensation and inhibition of DNA replication and RNA transcription. The small molecules bind to deoxyribonucleic acid (DNA) primarily in three modes: electrostatic interactions with the negative-charged nucleic sugar-phosphate structure, binding interactions with two grooves of DNA double helix and intercalation between stacked base pairs of native DNA [12]. MX recognizes chromatin structure with higher affinity than free DNA. Binding of MX to chromatin produces a very compact structure that prevents the release or extraction of histone proteins from such drug treated chromatin, its exact mode of interaction is still unknown [7].
Interactions of various anticancer drugs with DNA have been studied time and again by various workers using variety of techniques[13-16] and in recent years, the interest has grown in the electrochemical investigation of interaction between anticancer drugs and other DNA-targeted molecules and DNA [4,17-30]. A quantitative understanding of these interactions plays a significant role in recognition of DNA sites and would be valuable in the rational design of new DNA targeted molecules having wide applications in chemotherapy and in the development of tools of biotechnology based on DNA hybridization. Erdem et al [31] have electrochemically studied the interaction of MX with calf thymus double-stranded DNA (dsDNA) and calf thymus single-stranded DNA (ssDNA) using differential pulse voltametry (DPV) and cyclic voltametry (CV) at a carbon paste electrode (CPE) and observed that there were changes in MX signals, which are due to the interaction of MX with DNA, as decreasing signals. It was observed that the addition of an excess of dsDNA or ssDNA in MX solution caused changes in the peak current of the oxidation wave of MX. The concentration of MX also has a pronounced effect on its interactions with dsDNA. In both bare and dsDNA modified CPE’s, the response of MX increases sharply with concentration [29]. They further described the variations of voltametric behavior of MX in aqueous medium at a DNA modified CPE. It was done to modify promising DNA biosensors for a novel anticancer drug. DNA biosensors thus produced will eliminate the need for radioisotopes, as their hybridization time is significantly reduced as compared to counter radioisotopes. This determination of hybridization indicators which are capable of recognizing DNA would be valuable and open new horizons in the design of sequence-specific DNA binding molecules for application in chemotherapy and in the development of tools for the point-of-care tests and diagnosis based on DNA [31].
While taking into account the drug-DNA interactions the one of the important questions raised is the manner in which interaction takes place, is there any particular orientation of drug in which drug is introduced into the intercalating site or any sequence preference. The study of drug-DNA complexes carried by Ritu et al [10] showed that the MX interacts preferably in parallel mode, as the energy of interaction of MX in parallel mode is one order less than that of perpendicular mode. Also on binding in perpendicular mode the position of base pair is considerably altered. The conformation of side chains close to hydroxyl terminal is significantly different in two modes. Thus the orientation of ring system inside the intercalation site, the conformations of side chains and the conformation of DNA depend upon the relative position of substituent side chain. Therefore the knowledge of structure of drugs, their complexes with DNA and their correlation with anticancer activity have direct application in drug designing.
2.2 Sequence specificity shown by Mitoxantrone
To answer the query regarding preference of particular sequence by the drug, one of the first initiatives was taken by Kapuscinski et al [32]. On basis of the spectroscopic methods they concluded that there was “no clear base specificity” although interactions with polymers containing only A-T base pairs were somewhat weaker than with other polymers and alternating polymer exhibited stronger affinity than did homopolymer pairs. They restated this lack of significant DNA-base specificity of MX in another publication [33] in which they found a significant preference for the interaction with poly (dA-dT). Poly (dA-dT) rather than poly (dG-dC). Poly (dG-dC). Lown et al [34] in his work based on spectrophotometric methods, later obtained a conflicting result that both these agents show a preference for G-C sequence. This is particularly evident for MX whose binding constant to poly (dGdC). Poly (dG-dC) (= 47.5 х 10-5 M-1) was significantly larger than the poly (dA-dT). Poly (dAdT) (= 12.9 х 10-5 M-1) [35].
Further, both computer graphic studies [36] and a high-field 1H-NMR analysis of the interaction complex of MX with oligonucleotide duplex [37] showed that the preferential location for drug-DNA interaction is in the major groove, although studies with DNA in which major groove is partially blocked suggested that in such conditions “at least a part of the MX molecule may be in the major groove” [38]. The considerably less favorability of minor groove for MX binding than major groove is supported by many computer graphic studies [35]. In contrast to results obtained by computer graphic studies, the lack of reactivity of minor groove is not found due to the prohibitive repulsive interactions but instead to the lack of appropriate H-binding interactions emanating from MX towards the core of groove. The only H-bonding interactions present are those involving O1’ of S3 and S3’ sugars of the intercalating side with the side chain of MX. In another study carried out by Chen et al [8], it was observed that the best binding configuration of MX locates its two side chains in the major groove. A considerable preference is elicited for intercalation of the chromophore ring in a pyrimidine (3'-5') purine sequence rather than the isomeric purine (3'- 5') pyrimidine sequence. Contrary to the situation encountered with "simple" intercalations, in which this preference is generally attributed solely to differences in the energies of unstacking necessary to generate the intercalation sites, the preference is dictated in MX to a large extent by the intermolecular interaction energy term. This preference largely overcomes the base sequence preference as expressed in terms of G-C versus AT base pairs. Therefore it was concluded that most favorable intercalation sites for MX are those having 5’-purine base at the extremity of each strand and the best binding configuration involves the binding of the cationic side chains of O1 of the central phosphate group with N7 of the 5’-purine.
2.3 Mechanism of action
There is still a considerable degree of conjectures regarding the mechanism of action of MX. It has been found that mitoxantrone accumulates in cells and concentrates in the nucleus, where it interacts with nucleic acids but till date little is known about the mechanism by which MX binds to DNA. DNA is compacted into a complex structure built from the interaction of histones with DNA wrapped around an octamer of core histones. There are five main histones, one is linker histone of the H1 family and four core histones (H2A, H2B, H3 and H4) which are arranged in octamer form. Therefore the presence of chromosomal protein and the way in which they modulate binding of intercalating drug to DNA is important while trying to understand the mechanism of drug action at chromatin level [7]. From various studies it has been proved that MX intercalates into DNA and oxiditavely activated so that it can bind to nucleotides on DNA [11, 34, 39]. It has been found that activation of MX is carried out by oxidation of myeloperoxidase, the most abundant protein in neutrophil and is also found in monocytes [40]. Myeloperoxidase has subsequently been shown to oxidase MX to metabolites which bind covalently to DNA and RNA [41]. Since formaldehyde levels are known to be higher in cells of myeloid origin, this indicates a possible role of formaldehyde in the activation of MX. MX alone cannot form significant levels of interstrand cross links and therefore requires an extrinsic activation to enable covalent interaction with DNA [42]. In vitro studies revealed that the activation of MX by formaldehyde leads to the formation of drug-DNA adducts. It is presumed that drug binds to DNA via a methylene group provided by formaldehyde. The methylene group preferably associates with amino group. It associates on the alkyl amino side chain, forming an activated intermediate. Binding of formaldehyde to these sites form a Schiff base which is reactive and provides at least one potential binding site to nucleotides, yielding adducts [43]. It is formation of these adducts that stabilizes the DNA such that they functioned as virtual interstrand cross linkers. However it is possible that both amino groups on MX side chains might be activated, therefore potentially leading to a conventional interstrand cross links [42]. The formation of MX-DNA adducts in presence of myelperoxide and hydrogen peroxide is attributed to the oxidative activation of the drug [41]. Formation of interstrand cross links has also been shown in vitro by neutrophil activation of MX [40, 44]. In this case the formation of cross linkers was attributed to the increased amount of myeloperoxidase in the cells, but since these cells contain elevated levels of formaldehyde, this may account for activation of the drug and the subsequent binding to DNA. Konopa et al [45] reported DNA interstrand crosslink formation by MX in vivo.
Various studies were carried out time and again by researchers in order to gain understanding of the process of formation of adduct, investigating the role of formaldehyde in formation of DNA adduct and inter stand cross linked by MX in vitro. The results indicated that MX forms virtual DNA strand cross links both in vitro and in tumor cells and the formation of these lesions is dependent on both formaldehyde concentration, with cross linking being detected as low as 0.5 mM and on MX concentration, with cross linking being detected to < 5μM drug [42]. This MX concentration is similar to that used in clinic, where the dosage administered intravenously is 14 mg/m2 (~7μM). Moreover it has an effectively intracellular concentration that is approximately 80 fold more than the extracellular level [46]. The time dependent study of inter strand cross linking revealed that the reaction between MX and formaldehyde resulted in rapid binding of the drug to DNA, with the reaction reacting completion within 30 minutes at all concentrations studied. There is an increase in the generation of absolute level of inter strand cross linkers at higher concentrations of MX and formaldehyde [47].
It has also been found that MX activated by formaldehyde in vitro to from DNA adducts (Figure 2) are specific for CpG and CpA in DNA, and the stability of these adducts is dependent on flanking sequences. The most stable lesions detected to date are at ACGC sequences [48-49]. This specificity of CpG for adduct formation prompted investigations into the effect of cytosine methylation (CpG) on adduct formation, since the majority of CpG dinucleotide in the mammalian genome are methylated and hypermethylation in subsets of genes is associated with various neoplasms [50]. After carrying out the methylation of 512-base pair fragment (containing the lac UV5 promoter) using Hpa II methylase, it was observed that CCGG sites downstream of the promoter were methylated at C5 of the internal cytosine residue. In vitro transcription studies of MX-reacted DNA revealed that the transcriptional block frequency (hence adduct formation) was significantly enhanced (~ 3 fold), exclusively at the methylated sites. This finding was potentially important, since there is a high frequency of methylation at these sites in biological systems and is associated with differential localized hypoand hypermethylation in cancer cell lines [51-53]. Cross linking analysis assay also revealed that methylation enhanced duplex stabilization due to adduct formation by at least 3-fold at low formaldehyde concentration and 2-fold at higher concentration. Further adduct formation at a single50 potential drug binding site and the effect of methylation at this site was analyzed. It was observed that methylation enhanced adduct levels by 3-fold [50]. Collectively, these results indicate preferential adduct formation at methylated CpG sites. However, adduct at these methylated sites, exhibited the same stability as non-methylated sites, suggesting that cytosine methylation increases drug accessibility to DNA rather than being involved in kinetic stabilization of the adduct [54-55]. After knowing the importance of CpG island methylation on gene transcription, it has became easy to establish the ultimate effects of adducts at the site in terms of altered gene expressions.

Figure 2. Structure of mitoxantrone and DNA binding sites, A) mitoxantrone; B) proposed model of the formaldehyde-activated mitoxantrone-DNA monoadduct; C) a G-C base pair (where methylation of cytosine occurs at the C-5 position and projects into the major groove) (courtesy: B.S. Parker et al.2001.
Molecular modeling studies were also done to further understand the physicochemical interactions between MX and DNA [56]. Molecular geometries of MX and DNA bases (adenine, guanine, cytosine and thymine) were optimized and properties of isolated interface and its stacking interactions with adeninethymine (AT) and guanine-cytosine (GC) were studied. The B3LYP16-31G* stabilization energies of the interacalator-base pair complexes were found to be 10.06 Kcal/mol and 21.64 Kcal/mol for AT-MX and GC-MX respectively. It is concluded that the dispersion energy and electrostatic interaction contributed to the stability of the intercalator-DNA base pair complex.
3. Discussion
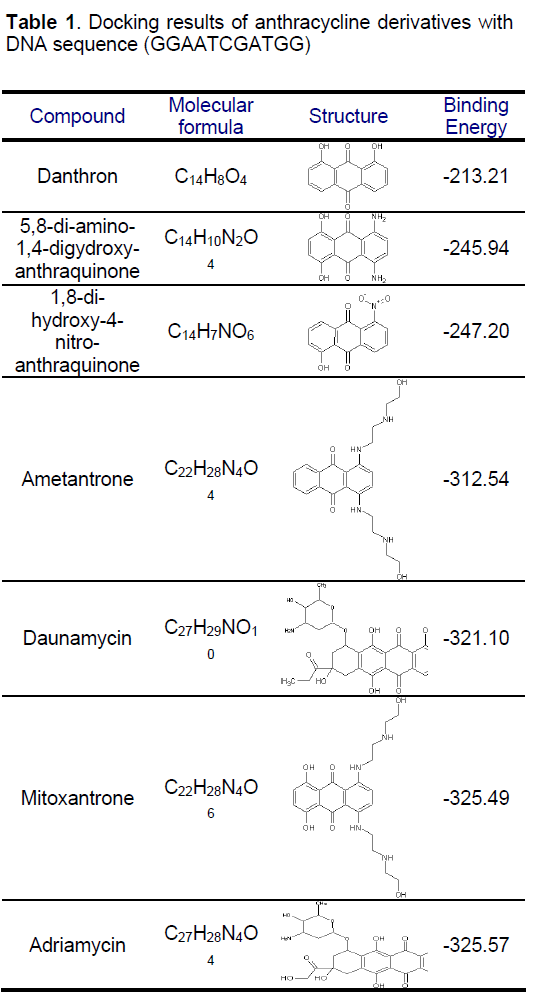
Mitoxantrone is an anthraquinone derivative with promising perspective in cancer chemotherapy. We docked different anthracycline derivatives into DNA sequence (GGAATCGATGG) using Hex 5.1 software [57]. The binding energy of mitoxantrone was found to be appreciably higher than some derivatives, while it was closer to known marketed drugs, adriyamycin and daunamycin (Table 1). The observation strengthened the hypothesis stating the comparable activity of mitoxantrone to the daunomycin and adriamycin, while being less cardiotoxic as MX do not reduces to semiquinone intermediate due to its more negative reduction potential than the anthracycline and therefore does not exhibit the cardio toxicity associated with the anthracyclines, andriamycin and daunomycin. Docking of MX into DNA sequence also confirmed the preferential binding of MX in the major groove of DNA (Figure 3) as compared to minor groove. Formaldehyde plays an important role in the activation of MX leading to the formation of Drug- DNA adducts. The positive correlation between cytotoxicity and methylation patterns could lead to the development of biological markers for potential sensitivity of individual patients to MX treatment. Severe binding of MX to histones implies that apart from DNA, histone proteins are also good candidate for MX action at chromatin level.

Figure 3. Mitoxantrone binding in major groove.
References
- DeVita V.T., Hellman S., Rosenberg S.A. (1993) Cancer: Principles and Practice of Oncology, 4th ed., J.B. Lippincott Co., Philadelphia, PA.
- Feofanov A., Sharonov S., Fleury F., Kudelina I., et al. (1997) Quantitative confocal spectral imaging analysis of mitoxantrone within living K562 cells: intracellular accumulation and distribution of monomers, aggregates, naphtoquinoxaline metabolite, and drugtarget complexes. Biophysics. J, 73: 3328-3336.
- Myers C.E., Mimnaugh E.G., Yeh G.C., et al. (1988) Anthracycline and Anthracenedione- based Anticancer Agents. Elsevier Amsterdam, 9: 527-569.
- Brett A.M.O., Macedo T.R.A., Raimundo D., et al. (1998) Voltammetric behavior of mitoxantrone at a DNA-biosensor. Biosensors and Bioelectron, 13: 861- 867.
- Cornbleet M.A., Stuart-Harris R.C., Smith I.E., et al. (1984) Mitoxantrone for the treatment of advance breast cancer: single-agent therapy in previously untreated patients. Eur. J. Cancer Oncol, 20: 1141- 1146.
- Pratt W.B., Ruddon R.W., Ensminger W.D., et al. (1994) In The Anticancer Drugs, 2nd Edn, Oxford University Press, New York.
- Zahra H., Azra R.C. (2009) Studies on the binding affinity of anticancer drug mitoxantrone to chromatin, DNA and histone proteins. J Biomed. Sci, 16: 31-36.
- Chen K.X., Gresh N., Pullman B. (1986) A theoretical investigation on the sequence selective binding of mitoxantrone to double-stranded tetranucleotide. Nucleic Acid Res, 14: 3799-3812.
- Lown J.W., Chen H., Plambeck J.A. (1982) Further studies on the generation of reactive oxygen species from activated anthracyclines and the relationship to cytotoxic action and cardiotoxic effects. Biochem. Pharmacol, 31: 575-581.
- Bhartwal R., Awasthi P., Kaur M., et al. (2003) Comparative study of intercalation complexes of mitoxantrone and ametantrone with d-CpG based on molecular dynamics approach. In National Symposium in Biophysics, NSB, IIT Roorkee.
- Reszka K., Hartley J.A., Kolodziejczyk P., et al. (1989) Interaction of the peroxidase-derived metabolite of mitoxantrone with nucleic acids: Evidence for covalent binding of 14C-labeled drug. Biochem. Pharmacol, 38: 4253-4260.
- Xia C., Guoli S., Jianhui J., et al. (1999) Intercalation of pharmorubicin anticancer drug to DNA studied by cyclic voltammetry with analytical applications. Anal. Lett, 32: 717-727.
- Norden B., Kubista M., Kurucsev T. (1992) Linear Dichroism spectroscopy of nucleic-acids. Quarterly Review of Biophysics, 25: 51-170.
- Lown J.W., Hanstock C.C., Bradley R.D., et al. (1984) Interactions of the antitumor agents mitoxantrone and bisantrene with deoxyribonucleic acids studied by electron microscopy. Mol. Pharm, 25: 178-184.
- Fritzsche H., Akhebat A., Taillandier E., et al. (1993) Structure and Drug interaction of parellel-stranded DNA studies by infrared spectroscope and fluorence. Nucl. Acids Res, 21: 5085-5091.
- Nunn C.M., Meervelt L.V., Zhang S. (1991) DNADrug Interactions: The Crystal Structures of d(TGTACA)and d(TGATCA) Complexed with Daunomycin. J. Mol. Biol, 222: 167-177.
- Wang J., Rivas G., Cai X., et al. (1997) DNA electrochemical biosensors for environmental monitoring. A review. Anal. Chim. Acta, 347: 1-8.
- Erdem A., Meric B., Kerman K., et al. (1999) Detection of interaction between metal complex indicator and DNA by using electro chemical biosensors. Electroanal, 11: 1372-1376.
- Erdem A., Kerman K., Meric B., et al. (1999) DNA Electrochemical Biosensor for the Detection of Short DNA Sequences Related to the Hepatitis B Virus. Electroanal. 11: 586-587.
- Erdem A., Kerman K., Meric B., et al. (2001) Novel hybridization indicator methylene blue for the electrochemical detection of short DNA sequences related to the hepatitis B virus. Anal. Chim. Acta, 422: 139-149.
- Fojta M., Doffkova R., Palecek E. (1995) Determination of traces of RNA in submicrogram amounts of single- or double-stranded DNAs by means of nucleic acid-modified electrodes. Electronal, 8: 420-426.
- Teijeiro C., Perez P., Marin D., et al. (1995) Cyclic voltammetry of mitomycin C and DNA. Bioelectrochem and Bioenerg, 38: 77-83.
- Marin D., Perez P., Teijeiro C., et al. (1998) Interactions of surface confined DNA with acidactivated mitomycin C. Biophysics. Chem, 75: 87-95.
- Wang J., Rivas G., Cai X., et al. (1990) Accumulation and trace measurements of phenothiazine drugs at DNA modified electrodes. Anal. Chim. Acta, 332: 139- 144.
- Wang J., Rivas G., Luo D., et al. (1996) DNAModified electrode for the Detection of Aromatic Amines. Anal. Chem, 68: 4365-4369.
- Wang J., Rivas G., Luo D., et al. (1996) DNA biosensor for the detection of hydrazines. Anal, Chem, 68: 2251-2254.
- Kelley S.O., Boon E.M., Barton J.K., et al. (1999) Single-base mismatch detection based on charge transduction through DNA. Nucl. Acids Res, 27: 4830- 4837.
- Kelley S.O., Jackson N.M., Hill M.G., et al. (1999) Long-Range Electron Transfer through DNA Films. Angew. Chem. Int. Ed, 38: 941-945.
- Plambeck J.A., Lown J.W. (1984) An Electrochemical Method of Measurement of the Binding of Doxorubicin and Danorubicin Derivatives to DNA. J. Electrochem. Soc.: Electrochem. Sci. and Tech, 131: 2556-2563.
- Marrazza G., Chiti G., Mascini M., et al. (2000) Detection of Human Apolipoprotein E Genotypes by DNA Electrochemical Biosensor Coupled with PCR. Clin. Chem, 46: 31-37.
- Erdem A., Ozsoz M. (2001) Voltammetry of the Anticancer Drug Mitoxantrone and DNA. Turk. J. Chem, 25: 469-475.
- Kapuscinski J., Darzynkiewicz Z., Traganos F., et al. (1981) Interactions of a new antitumor agent, 1,4- dihydroxy-5,8-bis2-(2-hydroxyethyl)amino- ethyl]amino-9,10-anthracenedione, with nucleic acids. Biochem Pharmacol, 30: 231–240.
- Kapuscinski J., Darzynkiewicz Z., Traganos F., et al. (1985) Interactions of antitumor agents ametantrone and mitoxantrone (novantrone) with double-stranded DNA. Biochem Pharmacol, 34: 4203-4213.
- Lown J.W., Hanstock C.C. (1985) High field 1H- NMR analysis of the 1:1 intercalation complex of the antitumor agent mitoxantrone and the DNA duplex d(CpGpCpG). J Biomol Struct Dyn, 2: 1097–1106.
- Foye W.O., Vajragupta O., Sengupta S.K. (1982) DNA-binding specificity and RNA polymerase inhibitory activity of bis(aminoalkyl)anthraquinones and bis(methylthio)vinylquinolinium iodides. J. Pharm. Sci, 71: 253–257.
- Patterson L.H. (1993) Rationale for the use of aliphatic N-oxides of cytotoxic anthraquinones as prodrug DNA binding agents: a new class of bioreductive agent. Cancer and Metastasis Reviews, 12: 119-134.
- Islam S.A., Neidle S., Gandecha B.M., et al. (1985) Comparative computer graphics and solution studies of the DNA interaction of substituted anthraquinones based on doxorubicin and mitoxantrone. J. Med. Chem, 28: 857–864.
- Lown J.W., Morgan A.R., Yen S.F., et al. (1985) Characteristics of the binding of the anticancer agents mitoxantrone and ametantrone and related structures to deoxyribonucleic acids. Biochemistry, 24: 4028- 4035.
- Mewes K., Blanz J., Ehninger G., et al. (1993) Cytochrome P-450-induced Cytotoxicity of MTX by formation of Elecrtophillic Intermediates. Cancer Res, 53: 5135-5142.
- Panousis C., Kettle A.L., Phillips D.R. (1994) Oxidative metabolism of mitoxantrone by the human neutrophil enzyme myeloperoxidase. Biochem. Pharmacol, 48: 2223-2230.
- Panousis C., Kettle A.L., Phillips D.R. (1995) Myeloperoxidase oxidizes mitoxantrone to metabolites which bind covalently to DNA and RNA. Anti-Cancer Drug Des, 593-605.
- Blenda S.P., Culliane C., Phillips D.R. (1999) Formation of DNA adducts by formaldehyde-activated mitoxantrone. Nucleic Acids Research, 27: 2918-2923.
- Taatjes D.J., Gaundiano G., Resing K., et al. (1997) A Redox Pathway Leading to the Alkylation of DNA by the Anthracycline, Anti-Tumor Drugs, Adriamycin and Daunomyci. J. Med. Chem, 40: 1276-1286.
- Panousis C., Kettle A.J., Phillips D.R. (1997) Neutrophil-mediated activation of mitoxantrone to metabolites which form adducts with DNA. Cancer Lett, 113: 173-178.
- Konoppa J. (1998) Molecular Aspects of Chemotherapy. Pergamon Press, New York, 83-94.
- Feofanov A., Sharonov S., Fleury F., et al. (1997) Localization and Molecular Interactions of MTX within living K562 cells as Probed by Confocal Spectral Imaging Analysis. J. Biophysics, 73: 3328- 3336.
- Perrin L.C., Cullinane C., Kimura K., et al. (1999) Barminomycin forms GC-specific adducts and virtual interstrand crosslinks with DNA. Nucleic Acid Res, 27: 1781-1787.
- Parker B.S., Cutts S.M., Cullinane C., et al. (2000) Formaldehyde activation of mitoxantrone yields CpG and CpA specific DNA adducts. Nucleic Acids Res, 28: 982-990.
- Parker B.S., Cutts S.M., Phillips D.R. (2001) Cytosine Methylation Enhances Mitoxantrone-DNA Adduct Formation at CpG Dinucleotides. J.Biol.Chem, 276: 15953-15960.
- Parker B.S., Buley T., Evison B.J., et al. (2004) Cytosine Methylation Enhances Mitoxantrone-DNA aduct Formation at CpG Dinucleotides. J. Biol. Chem, 279: 18814-18823.
- Baylin S.B., Hoppener J.W.M., Bustros A.D., et al. (1986) DNA Methylation Patterns of the Caleitonin Gene in Human Lung Cancer and Lymphomas. Cancer Res, 46: 2917-2922.
- Villian A., Vogt N., Dutrillaux B., et al. (1999) DNA methylation and chromosome instability in breast cancer cell lines. FEBS Lett, 460: 231-234.
- Melki J.R., Vincent P.C., Clark S.J. (1999) Concurrent DNA Hypermethylation of Multiple Genes in Acute Myeloid Leukemia. Cancer Res, 59: 3730-3740.
- Millard J.T., Beachy T.M. (1993) Cytosine methylation enhances mitomycin C cross-linking. Biochemistry, 32: 12850-12856.
- Denissenko M.F., Chen J.X., Tang M.S. (1997) Cytosine methylation determines hot spots of DNA damage in human P53 gene. Proc. Natl. Acad. Sci, 94: 3893-3898.
- Riahi S., Ganjali R.M., Dinarvand R., et al. (2008) A theoretical study on interaction between MTX as an anticancer drug and DNA: application in drug design. Chemical Biology and Drug Design, 71(5): 474-482.
- URL: https://www.loria.fr/~ritchied/hex/


Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences