How Far is ITS2 Reliable as a Phylogenetic Marker for the Mosquito genera?
Amit Kumar Banerjee, Neelima Arora, Upadhyayula S.N. Murty
Amit Kumar Banerjee,Neelima Arora,Upadhyayula S.N. Murty*
Bioinformatics Group,Biology Division,Indian Institute of Chemical Technology,Tarnaka,Hyderabad-500007,Andhra Pradesh,India
Abstract
The present study provides substantial evidence of variation among the ITS2 regions of the sample species both at sequence as well as RNA secondary structure level. A bioinformatics approach was exploited to reveal the mystery of conservation and variation in ITS2 in mosquito genera. Multiple sequence alignment revealed conservation within the species and across species. Dimer frequency of all the species showed variance in conservation according to the type of dimer. RNA secondary structures were generated for all the species. To obtain a deep insight into structural similarity and divergence, parameters like structural energy, number of stems, G-C, A-U, G-U pairings were calculated. Out of five types of loops derived by Sribo Program, hairpin and exterior loops were found to be fairly more conserved rather than interior, bulge and multi loops. Tandem repeats with high degree of variation were observed in these specific regions. The unexpected observation was the striking proximity of Aedes longfilamentus and Anopheles culicifacies in the phylogenetic tree where they shared same clade instead of other species of Aedes genera. This finding is quite appealing and intriguing being contrary to the expected outcome and requires further rigorous investigation. These results indicate towards the major variable and conserved regions as well as the stretches of sequences and structural parts where selection pressure varies during ribosome biogenesis.
Keywords
ITS2,Phylogenetic marker,RNA Secondary structure,Mosquito,Anopheles,Aedes.
1. Introduction
Perilous mosquito-borne diseases like Malaria,Chikungunya and Dengue put a massive social and economic burden globally. In the wake of reemergence of mosquito-borne diseases,studies at the molecular level have gained momentum during past decades. Out of 450 Anopheline species known,only 30-40 species act as vectors for malaria while the proportion of Aedes species acting as vector is still less in comparison. Anopheles and Aedes mosquitoes are extensively distributed throughout the globe [1]. An. culicifacies,An. fluviatilis,An. minimus and An. sundaicus are predominant in the Indian peninsular region. Anopheles farauti,An. gambiae and An. darlingi are widely spread in Australia,Africa and Latin America respectively. Anopheles nuneztovari is predominant in diverse geographical locations of Latin America [2]. Aedes species are also distributed through out the world and lot of studies have focused on the taxonomy of these species globally in the recent past [3].
1.1 Internal Transcribed Spacer 2 (ITS2)
Two internal transcribed spacers (ITS) occur in eukaryotic organisms namely ITS1 and ITS2. ITS1 and ITS2 are flanked by 18S gene and 5.8S gene,5.8S gene and 28S gene respectively [4]. ITS2 is regarded to be more conserved than ITS1 owing to presence of numerous tandem repeats. The ITS2 region is now the most widely sequenced DNA region in mosquito genera of Anopheles,Culex and Aedes [5]. Complete ITS2 sequences of nine species each of Anopheles and Aedes genera from diverse geographical locations were considered for this study.
1.2 ITS2 as a phylogenetic marker in mosquitoes
The ITS2 portion of ribosomal DNA is considered as extremely conserved and species-specific. Among the hugely considered molecular markers for mosquito taxonomy [5-6],ITS2,part of ribosomal DNA,is exploited extensively for distinguishing the closely-related species. This marker is extensively used in Anopheline complexes like,An. maculipennis complex,An. quadrimaculatus complex [7] and An. culicifacies complex [8]. Research targeting the ITS2 region for species classification and phylogenetic analysis is gaining impetus globally [6] [9]. In the last decade,ITS2 regions of several Neotropical Anophelines have been sequenced and dumped in the GenBank database. Therefore,the conservation and variation comparison of the ITS2 sequences across the species as well as genera is the prime objective of this study.
2. Materials and Methods
2.1. Data set
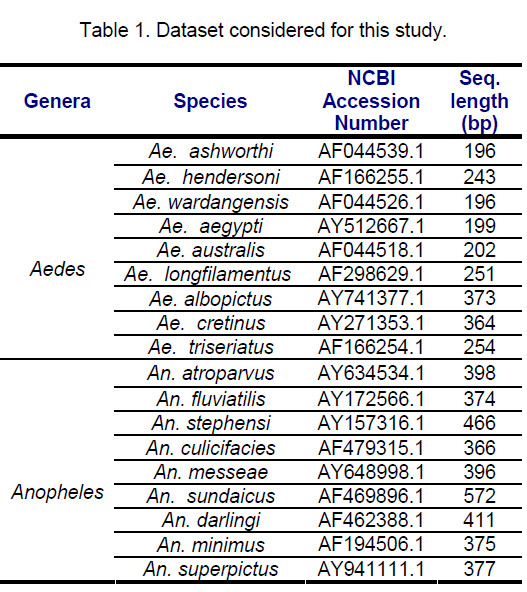
ITS 2 sequences of 18 mosquito species belonging to diverse geographical locations,9 each from Aedes and Anopheles genera,retrieved from Genbank were investigated. The species considered for this study along with the NCBI accession number and lengths (bp) are shown in the table below (Table 1).
2.2 Sequence alignment
Multiple sequence alignment was performed using CLUSTALW [10] with a gap opening penalty of 15 and gap extension penalty of 6.66. The result of ClustalW analysis is graphically represented by using GeneBee server [11] (Figure 1-3).

Figure 1. Alignment of ITS2 sequences of Anopheles species.

Figure 2.Alignment of ITS2 sequences of Aedes species.

Figure 3.Alignment of ITS2 sequences of all 18 species.
2.3 Reconstruction of Phylogeny
The phylogenetic tree was constructed considering the ITS2 region by using the program PHYLOWIN [12].196 sites were generated. Other parameter considered was divergence observed with 500 bootstrap replicates. The tree was built using Neighbor-joining method. The consensus tree along with branch distances and bootstrap number is shown in Figure 4 .

Figure 4.Phylogenetic tree of the 18 mosquito species based on the ITS2 sequences.
2.4 Calculation of tandem repeats
All the species were considered for locating tandem repeats. Tandem Repeats Occurrence Locator (TROLL) [13] was used for determining the conserved repeats in the considered ITS2 sequences. The maximum motif length considered was 6 and the minimum repeat length was 10.The observed results are tabulated in Table 2.

2.5 Microsatellite analysis
Frequency of the bases is an important measure for sequence conservation analysis. Dimer frequency was calculated using Spectrum repeat finder (SRF) [14]. The most frequent dimer repeats and the extended range of their distribution in all the considered ITS2 sequence was calculated using SRF. Obtained results are shown in Figure 5 -8.

Figure 5.Species-wise distribution of CG repeat and the spanned sequence stretch.

Figure 6.Species-wise distribution of dimer frequency.

Figure 7.Species-wise distribution of CA repeat and the spanned sequence stretch.

Figure 8.Species-wise distribution of dimer frequency other than CA repeats.

Figure 9.Comparison of different types of loops of Anopheles Internal Transcribed Spacer2.
2.6 Secondary structure prediction
RNA secondary structure consists of stems and loops. Mainly five types of loops are present in RNA secondary structure,namely,interior,hairpin,exterior,multi and bulge. For in-depth analysis,calculation of secondary structure and determination of structural conservation is essential.
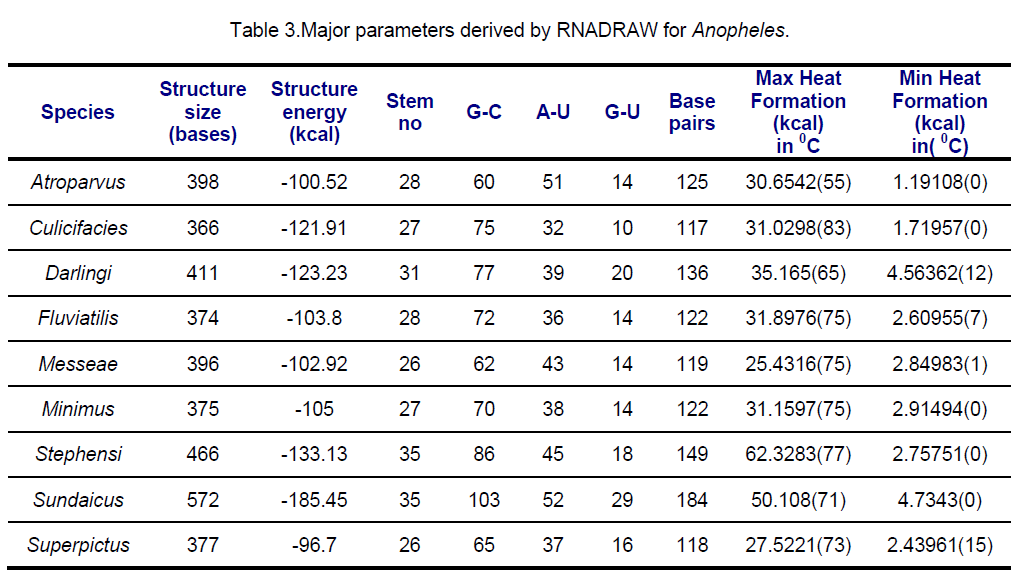
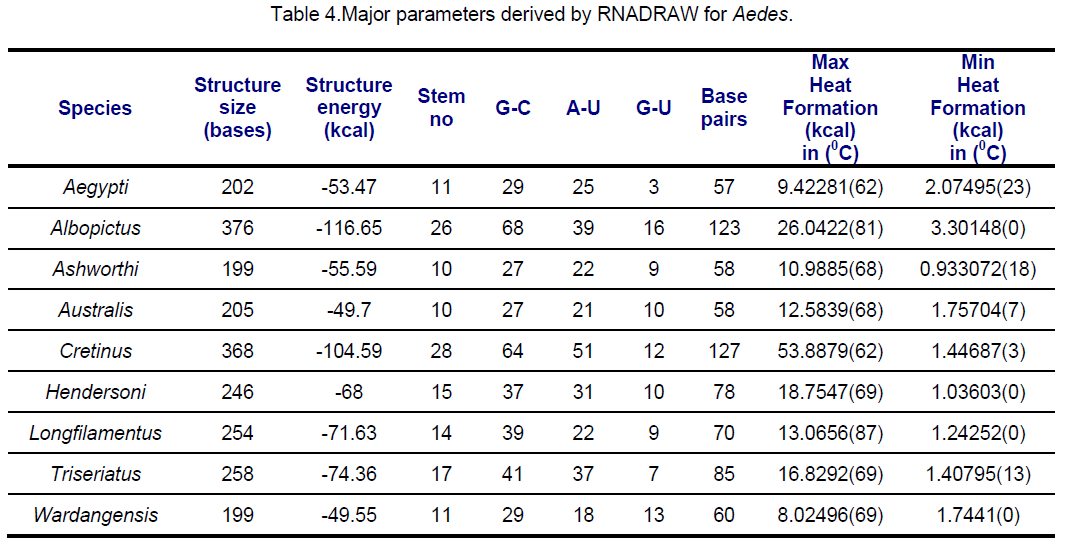
RNA secondary structures for ITS2 were predicted using RNADRAW [15-16] program. The dynamic programming algorithm utilized in RNADRAW is based on the effort of Zuker and Stiegler [17] and employs energy parameters taken from Freier [18] and Jaeger [19]. In the current study,complete sequences of ITS2 were used for RNA structure prediction. The minimum energy structure prediction algorithm in RNADRAW was ported from the RNAFOLD program included in the Vienna RNA package. Obtained results are shown in Table 3,4.
2.7 RNA folds analysis
Based on a statistical sample of the Boltzmann ensemble for secondary structures,Sribo program in Sfold (statistical Folding and Rational Design of Nucleic Acids) [20] was used to predict the probable target accessibility sites (loops) for trans-cleaving ribozymes in ITS2. Here,the likelihood of unpaired sites for potential ribozyme target was assessed. Due to existence of RNA as a population of different structures,stochastic approach to the evaluation of accessible sites was found to be appropriate [15]. The probability profiling approach by Ding and Lawrence [21] reveals target sites that are commonly accessible for a large number of statistically representative structures in the target RNA,thus bypassing the long-standing difficulty in accessibility evaluation due to limited representation of probable structures owing to high statistical confidence in predictions. The probability profile for individual bases (W=1) was produced for the region that includes a triplet and two flanking sequences of 15 bases each in every site of the selected cleavage triplet. Figure 9 and 10 shows the observed results.

Figure 10.Comparison of different types of loops of Aedes Internal Transcribed Spacer 2.
3. Results and Discussion
ITS2 length varies from 374- 572 bp in Anopheline species where An. fluviatilis and An. minimus showed similar length. The length variations were observed with maximum length being 374 bp and minimum of 572 bp for An.fluviatilis and An. sundaicus respectively. ITS2 in Aedes showed comparatively smaller size of ITS2 sequences that ranged from 196- 373 bp with Aedes ashworthi showing the shortest ITS2.
The ClustalW results show the expected better alignment in the Anopheles and Aedes genera individually as well as dissimilarity when all the 18 species of both the genera were considered simulataneously (Figure 1,2). GC content among Aedes species varied between 48.7 and 55.5% while in considered Anopheline species,it ranged from 51.5 to 59.4%.
The multiple sequence alignments in Figure 1 and Figure 2 depicted the variation in the alignment very clearly. Species considered from Aedes genera showed better conservation at the sequence level than Anopheles genera. The overall alignment of the 18 species shows better alignment from position 150 to 320 (Figure 3 ).
The lowest distance among all the sequences is observed in between Anopheles minimus and An. fluviatilis. An. culicifacies and Aedes longfilamentus share the same node with an observed divergence of 0.1 distance.
The tandem repeat search revealed variations. Some of the species in both the genera do not show any tandem repeats (Ae. australis,Ae. ashworthi,Ae. wardangensis,An. atroparvus,An. messeae,An. culicifacies,An. minimus,An. sundaicus ) in the constraint range.
TC and GC repeat are same in case of Ae.australis. Same dimer frequency for these repeats among all the 3 species is another interesting feature observed. Ae. australis and Ae. ashworthi also show same GC repeat number. Dimer frequency varies from 16 to 33 in case of CG repeats but as far as other dimers are concerned,they maintain the same copy number. Ae.triseriatus,Ae. hendersoni and Ae. longfilamentous are found to be having same copy number in the same repeat searching region. Aedes aegypti and Ae. albopictus also possess same copy number in their spanning region.
CA repeats are found to have same copy number in An. messeae and An. stephensi are found even though region ranges are different.
TG repeats are found to be almost same in An. fluviatilis,An. minimus but differ significantly in An. sundaicus. Difference in GT repeats is observed between An. superpictus and An. sundaicus.
Secondary structural features of ITS2 regions are presented in Table 3,4.
ITS2 RNA structures from An.sundaicus has the highest negative free energy (-185.45 Kcal) followed by An. stephensi (-133.13 Kcal) while An.superpictus showed lowest negative Structural energy. Structural energy is found to be same in An. fluviatis and An. messeae and quite same in An. culicifacies and An. darlingi.. Surprisingly,GU pairing is same in 4 species viz. An.atroparvus,An.fluviatilis,An.messeae and An. minimus. Similar maximum heat formation is observed in An. culicifacies,An.fluviatilis,An. minimus.
Highest negative structural energy was observed in Aedes albopictus (-116.65 Kcal) followed by Ae.cretinus (-104.59) while Ae. wardangensis possess lowest negative energy. Structural energy is found to be similar in Ae. australis and Ae. wardangensis viz -49.7 and -49.5 respectively and it varies in trivial manner in Ae. aegypti and Ae. ashworthi.
The stems (double stranded paired regions) stabilize RNA secondary structure. The Anopheles species considered in this study were grouped into different classes based on the similarity of RNA stems and loops. Class I comprising (An. stephensi and An. sundaicus) showed similarity in secondary structural features. Similar observations were found in Class II (An.messae and An. superpictus) and Class IV (An.atroparvus and An. fluviatilis).Despite the different geographical regions,An. culicifacies and An. minimus are falling in the class III sharing similar secondary structural features.
Similarly in Aedes,ITS2 were classified into 3 classes; Class I (Ae. wardangensis and Ae.aegypti),Class II (Ae. ashworthi and Ae.australis) based on similar structural properties while rest others showing high variance in structural features were clubbed as Class III. An interesting observation was that on visual comparison of secondary structure with the phylogenetic tree,Anopheles atroparvus,An. fluviatilis and Aedes cretinus which share the same stem number (28) were found close to each other in the tree too.
This convergence at secondary structural level among few species from different regions may be due to the evolutionary pressure on ITS2 to maintain the RNA secondary structure involved in post-transcriptional processing of rRNA.
The order of preference for conservation is observed to be more in the case of hairpin loop followed by exterior and bulge loop but surprisingly considerable variation is present in the interior loop type. In exterior loop and multi loop,An. darlingi shows significant variation to other species. In Ae. ashworthi,no bulge loop is observed. Maximum conservation among species is observed in exterior loop followed by hairpin loop while maximum variation is found in interior loop
Wesson et al [22] had described several homologous domains in the ITS regions of Aedes mosquito species and these domains are known to undergo base pairing to form a core region crucial to various stem attributes. This suggested the role of conserved core region of rDNA in functional rRNA folding pattern. ITS2 is required for the precise and efficient processing and maturation of 26S rRNA ribosomal units [23]. The distribution of information needed for the efficient removal of ITS2 from its RNA precursor is not localized but scattered throughout the ITS2 region. Variation and changes in rRNA folding pattern due to evolutionary sequence variation in the ITS spacer regions may have an imperative role on the ribosomal biogenesis.
4. Conclusion
ITS2,a well-known phylogenetic marker,is studied for contrasting features that form the basis of evolutionary process i.e. the general trend of variability among the species as well as the conservation among few species. At the sequence level and in loop regions,Aedes is found to show less variation with respect to the Anopheles species. Results of the present study suggests that exterior loops are the most conserved types of loops followed by hairpin,bulge and multi loops .But Aedes has shown similarity with Anopheles in secondary structure features by showing maximum divergence in interior loop conservation. Identifying the homologous regions and reconstructing their evolution increases the traits available for the phylogenetic analysis. Construction of an evolutionary tree using more species will provide an understanding for their functional selection. The present study indicates the phylogenetic relationships among the mosquito species belonging to different genera. In nutshell,this study shows that Aedes ITS2 region is more conserved than Anopheles ITS2. This indicates a possible role of varied selection pressure in this variation and it may be a plausible cause of greater divergence of Anopheles rather than Aedes species. This study raises a question on reliability of ITS2 in Anopheles as a phylogenetic marker as compared to Aedes. Further rigorous investigation is required considering all the species of Anopheles and Aedes to prove the reliability of ITS2 as a phylogenetic marker in context of mosquito species. New neutral and efficient molecular markers are required to study the origin and phylogeny of mosquito.
Acknowledgements
The authors are grateful to Dr. J.S. Yadav,Director,IICT,India for his constant support and encouragement. Neelima Arora thanks CSIR for Senior Research Fellowship.
References
- Kiszewksi et al. (2004) Global distribution (Robinson projection) of dominant or potentially important malaria vectors. American Journal of Tropical Medicine and Hygiene,70(5):486-498.
- Fritz,G.N.,Conn,J.,Cockburn,A.F.,et al. (1994) Sequence Analysis of the Ribosomal DNA Internal Transcribed Spacer 2 from Populations of Anopheles nune~tovari (Diptera: Culicidae). Molecular Biology and Evolution,11: 406-416.
- Mahadev,P.V.M.,Fulmali,P.V.,Mishra,A.C. (2004) A preliminary study of multilevel geographic distribution & prevalence of Aedes aegypti (Diptera: Culicidae) in the state of Goa,India. Indian J Med Res,120: 173-182.
- Perry,R.P. (1976) Processing of RNA. Annu. Rev. Biochem,45: 605-629.
- Wesson,D.M.,Porter,C.H.,Collins,F.H. (1992) Sequence and secondary structure comparisons of ITS rDNA in mosquitoes (Diptera: Culicidae). Molecular phylogenetics and evolution,1: 253-2699.
- Marrelli,M.T.,Sallum,M.A.M.,Marinotti,O. (2006) The second internal transcribed spacer of nuclear ribosomal DNA as a tool for Latin American anopheline taxonomy -A critical review. Mem Inst Oswaldo Cruz,Rio de Janeiro,101(8): 817-832.
- Cornel,A.J.,Porter,C.H.,Collins,F.H. (1996) Polymerase chain reaction species diagnostic assay for Anopheles quadrimaculatus cryptic species (Diptera: Culicidae) based on ribosomal DNA ITS2 sequences. J Med Entomol,33: 109-116.
- Goswami,G.,Raghavendra,K.,Nanda,N.,et al. (2005) PCR-RFLP of mitochondrial cytochrome oxidase subunit II and ITS of ribosomal DNA: markers for the identification of members of the An. culicifacies complex (Diptera :Culicidae). Acta Trop,95(2): 92-99.
- Severini,C.,Silvestrini,F.,Mancini,P.,et al. (1996) Sequence and secondary structure of the rDNA second internal transcribed spacer in the sibling species Culex pipiens L. and Cx. quinquefasciatus Say (Diptera: Culicidae). Insect Mol. Biology,5(3): 181-186.
- Higgins,D.,Thompson,J.,Gibson,T. (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting,position-specific gap penalties and weight matrix choice. Nucleic Acids Res.,22: 4673-4680.
- Brodsky,L.I.,Vasiliev,A.V.,Kalaidzidis,Ya. L.,et al. (1992) GeneBee: the program package for biopolymer structure analysis. Dimacs,8: 127-139.
- Galtier,N.,Gouy,M.,Gautier,C. (1996) SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci,12: 543-548.
- Castelo,A.,Martins,W.,Gao,G. (2002) TROLL-Tandem Repeat Ocurrence Locator. Bioinformatics,18(4): 634-636.
- Deepak,S,Biju,I.,Raghava,G.P. S.,et al. (2004) Spectral Repeat Finder (SRF): identification of repetitive sequences using Fourier transformation. Bioinformatics,20(9): 1405-1412.
- Christoffersen,R.E.,McSwiggen,J.A.,Konings,D. (1994) Application of computational technologies to ribozyme biotechnology products. J. Mol. Structure (Theochem),311: 273-284.
- Hofacker,I.L.,Fontana,W.,Stadler,P.F.,et al. (1994) Fast Folding and Comparison of RNA Secondary Structures. Monatsh. Chem.,125: 167-188.
- Zuker,M.,Stiegler,P. (1981) Optimal computer folding of large RNA sequences using thermodynamics and auxiliary information. Nucl. Acids. Res.,9: 133-148.
- Freier,S.M.,Kierzek,R.,Jaeger,J.A.,et al. (1986) Improved free-energy parameters for predictions of RNA duplex stability. Proc. Nat. Acad. Sci. U S A,83(24): 9373-9377.
- Jaeger,J.A.,Turner,D.H.,Zuker,M. (1989) Improved predictions of secondary structures for RNA. Proc. Natl. Acad. Sci. U S A,86(20):7706-7710.
- Ding,Y.,Chan,C.Y.,Lawrence,C.E. (2004) Sfold web server for statistical folding and rational design of nucleic acids. Nucleic Acids Res.,32(Web Server issue): 135-141.
- Ding,Y.,Lawrence,C.E. (2001) Statistical prediction of single-stranded regions in RNA secondary structure and application to predicting effective antisense target sites and beyond. Nucleic Acids Res.,29: 1034-1046.
- Wesson,D.M.,Porter,C.H.,Collins,F.H. (1992) Sequence and secondary structure comparisons of ITS rDNA in mosquitoes (Diptera: Culicidae). Mol. Phylogenet. Evolution,1(4):253-69.
- Van der Sande,C.A.,Kwa,M.,Van Nues,R.W.,et al. (1992) Functional analysis of internal transcribed spacer 2 of Saccharomyces cerevisiae ribosomal DNA. J.Mol.Biol. 223(4):899-910.


Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences