Direct Electron Transfer of Hemoglobin on Cadmium Oxide Nanoparticles Modified Carbon Paste Electrode
Gh. Mazaheri, M. Fazilati, S. Rezaei-zarchi, M. Negahdary, A. Kalantar-Dehnavy, M. R. Hadi
1Department of Biology,Payam-e-Noor University,Tehran,Iran.
2Department of Biology,Payam-e-Noor University,Isfahan,Iran.
3Department of Biology,Payam-e-Noor University,Yazd,Iran
4Department of Biology,University of Shahr-e-kord,Shahr-e-kord,Iran.
- Corresponding Author:
- Gh. Mazaheri
Tel:+98-913-332-7650
Fax: +98-332-322-9650
E-mail: gmazaheri@ymail.com
Abstract
Direct electron transfer of hemoglobin, immobilized on a cadmium oxide nanoparticles modified carbon paste electrode, was investigated. Cadmium oxide nanoparticles synthesized by chemical methods. The prepared nanoparticles were characterized by scanning electron microscope (SEM) and transmission electron microscope (TEM). The resulting electrode displayed a superior redox behavior for the hemoglobin. The hemoglobin showed a quasi-reversible electrochemical redox behavior with a formal potential of -50 mV (versus SCE) in 50 mM potassium phosphate buffer solution at pH 7.0 and temperature 25°C. The cathodic transfer coefficient was 0.40 and electron transfer rate constant was evaluated to be 1.47 s−1. Moreover, the modified electrode was used as a biosensor and exhibited a satisfactory stability and sensitivity to NO. In addition, NO induced a cathodic potential shift of the catalytic reduce peak of oxygen. This potential shift was proportional to the logarithm of NO concentration ranging from 5.0 × 10−8 to 5.0 × 10−6 mol/L. The detection limit has been estimated to be 50 nM. Thus the linear range of this biosensor for NO determination was from 0.05 to 5 μM while standard deviation in 5 μM NO concentration was 2.5% for 4 repetitions
Keywords
Electron transfer,hemoglobin,cadmium oxide nanoparticles,biosensor,NO.
1. Introduction
The alteration of electrode surface with the use of nanostructure materials as a mediator,in a word nanofabrication,is advantageous for the achievement of direct electron transfer (ET) between the biomolecule and the electrode [1]. The direct ET can be difficult to attain,because the prosthetic groups of the biomolecules are burried deeply in the biomolecules [2]. Nanomaterials are proportionate in size to proteins and the multivalent functionalization on their surfaces holds great promise for controlling the biomolecular recognition [3].
Direct ET has extended beyond the field of bioelectrochemistry. It is a new interdisciplinary area,which combines biotechnology with electrochemical science and focuses on the structural organization and electron transfer functions of biointerfaces on electrode surfaces [4]. Bioelectrochemistry has proven to be both a useful way to understand the electrochemical properties and principles of biomolecules as well as a powerful method for the exploitation of these biomolecules at the biointerfaces of biosensors and bioelectronics. Since that time,electrochemical devices have opened up new possibilities for studying the redox process of heme proteins. Many reports have described the electrochemistry of heme proteins in terms of modifier-electrode and modifier-protein interactions [5]. Many promoters,like some small organic compounds,amino acids together with some derived molecules,small peptides and conductive polymers,have been found to promote the direct electrochemistry of heme proteins on the electrode surface. Recently,the matrices used for the heme proteins immobilization are a series of inorganic porous and nanomaterials [3,5]. The study of direct electron transfer between protein and electrode represents not only a basic feature for the application of biocatalysts in chemical sensors and other electrochemical devices,but may also provide a model for the investigation of electron transfer processes in biological systems [3]. In previous studies,it had also revealed that the nanosilver facilitates electron transfer between hemoglobin and the surface of graphite electrode [6].
Here,in this study,we investigate the electrochemical behavior of hemoglobin in the presence of cadmium oxide nanoparticle modified carbon paste electrode. Modified carbon paste electrode surface with cadmium oxide nanoparticles,the ability for hemoglobin sensing was made possible. This sensing can be used to design biosensor to measure NO gas,because NO has a high affinity to heme iron of hemoglobin,and electrochemical and biochemical studies have revealed that NO penetration into the pocket of the hemoglobin disrupts the coordination bond between ferrous heme and O2,which finally produces met-Hb. Therefore,it is anticipated that traces of NO in solution may compete with O2,and the electrocatalysis behavior of hemoglobin reduction is modulated,while of this property can use to measure NO in solution. Hb immobilized on the electrode surface facilitates the reduction of oxygen,and indeed catalyzes its reduction [7].
2. Experiment
Reagents
Hemoglobin (Hb) was obtained from Sigma,USA,and used without further purification. Cetyl trimethyl ammonium bromide (CTAB) was purchased from Sigma,too. The phosphate buffer solution (PBS) consisted of a potassium phosphate solution (KH2PO4 and K2HPO4 from Merck,0.05 mol L-1 total phosphate) at pH 7.0. An acetate buffer solution (CH3COONa and CH3COOH from Merck,0.10mol L-1) was freshly prepared. Cadmium sulphate and Sodium hydroxide were from S.D fine chemicals. Potassium nitrite and other materials purchased from Merck. Deionized water was used to prepare all solutions and to rinse the electrodes double distilled water was used. Stock solutions were stored at 4°C.
Apparatus and measurements
Electrochemical measurements were carried out with a potentiostat/galvanostat (Palm-Sens,Netherland),equipped with a personal computer and treating of data. A conventional three electrode cell was employed throughout the experiments,with bare or cadmium oxide nanoparticles modified carbon paste electrode (3.0 mm diameter) as a working electrode,and all potentials reported here were referred to this electrode,a saturated calomel electrode (SCE) as a reference electrode which all potentials were reported with respect to this reference,and a platinum electrode as a counter electrode. All the electrochemical measurements were carried out in 0.05 M PBS,pH 7.0,at 25 ± 0.5 °C. Scanning electron microscopic images were recorded using a ZEISS DSM 960,while transmission electron microscopic studies were performed with the help of a ZEISS CEM 902A. X-ray diffraction of CdO nanoparticles examined by a Brucker D/MAX 2500 X-ray diffractometer with Cu Kα radiation (λ=1.540 56 Å),and the operation voltage and current were maintained at 40kV and 250mA,respectively. UV–VIS absorption spectroscopy was performed at a UV-2201 spectrophotometer (Shimadzu,Japan).
Preparation of CdO nanoparticles
In a typical experiment first solution prepared using 0.03M CdSO4,0.06M CH3COOH and 40 mg CTAB as surfactant in 1 dm3 of double distilled water. The second solution was prepared by 0.09 M NaOH pallets and 25ml 70% ethanol in 1 dm3of double distilled water. Then first solution was added to second solution with continues stirring. The obtained precipitate was filtered by using Whatmann filter paper (grade-41) and dried at 80°C in hot air oven about 1hour. Then dried precipitate was transferred to silica crucible and ignited at 400°C for about 4hours. Then obtained powder was washed with ethanol three to four times to remove impurities present in the particles. Then these are characterized using XRD,UV-Visible absorption Spectroscopy,and applied for fabrication of carbon paste electrode for determination of NO.
Preparation of bare carbon paste electrode
The bare carbon paste electrode was prepared by hand mixing of 70% graphite powder with 30% silicon oil in an agate mortar to produce a homogenous carbon paste. The paste was packed into the homemade cavity (3.0 mm in diameter) and then smoothed on a weighing paper. The electrical contact was provided by a copper wire connected to the paste in the end of the tube.
Preparation of CdO nanoparticles modified carbon paste electrode
The CdO nanoparticles modified carbon paste electrode was prepared by hand mixing of 70% graphite powder and 10 mg CdO nanoparticle with 30% silicon oil in an agate mortar to produce a homogenous carbon paste. The paste was packed into the homemade cavity (3 mm in diameter) and then smoothed on a weighing paper. The electrical contact was provided by a copper wire connected to the paste in the end of the tube.
Preparation of Hb/cadmium oxide NP/ carbon paste electrode
The prepared CdO NPs/ carbon paste electrode was placed into a fresh PBS including 3 mg mL-1 Hb (pH 7.0,3 to 5°C) for 8 h. At the end,the modified electrode was washed in deionized water and placed in PBS (PH 7.0) at a refrigerator (3 to 5°C),before being employed in the electrochemical measurements as the working electrode.
Preparation of saturated solution of NO
Asaturated solution of NO was obtained by treating 50 g of KNO2 with 125 mL of 6 M H2SO4 according to the following equation: 3KNO2 + H2SO4 = 2NO + KNO3 + K2SO4 +H2O. The resulting NO was purified by passing through three separate trap units of 3mol/L NaOH twice and distilled water once to eliminate contaminations. Then it was saturated in 0.1M phosphate buffer (pH 7.4) and the final concentration of NO was 2.0mM. Dilution of this stock NO solution was freshly made to prepare standard NO solution of different concentrations.
3. Results and discussion
Figure 1 shows the SEM image of synthesized cadmium oxide nanoparticles. This image shows the particle size of CdO in the nanometer range.

Figure 1: SEM image of CdO.
The XRD pattern for CdO nanoparticles,was shown in figure2. The diffraction peaks are absorbed at 2θ values. The prominent peaks have been utilized to estimate the grain size of sample with the help of Scherrer equation D = Kλ / (β cosθ) where K is constant (0.9),λ is the wavelength (λ = 1.5418 A°) (Cu Kα),β is the full width at the half-maximum of the line,D is the average crystallite grain size and θ is the diffraction angle. The grain size estimated using the relative intensity peak (100) for CdO nanoparticles was found to be 45 nm and increase in sharpness of XRD peaks indicates that particles are in crystalline nature.

Figure 2: XRD pattern for CdO nanoparticles.
Figure 3 a shows the TEM image of carbon paste electrode surface before the construction of cadmium oxide nanoparticles. Figure 3 b shows the TEM images of cadmium oxide nanoparticles after being scraped from the electrode surface. Because the surface-to-volume ratio increases with the decreasing size,the smaller NPs are able to play a very important role during the immobilization process.

Figure 3: TEM images of bare carbon paste electrode (a), and after modified with CdO NPs (b).
The electron transfer of the proteins,at the bare electrodes,is very slow so that the redox peak of proteins can usually not be observed [8]. Figure 4 a shows a typical cyclic voltammogram (CV) of the CdO-NPs/ carbon paste electrode. Figure 4 b shows a cyclic voltammogram of an Hb/CdO-NPs/ carbon paste electrode in 50 mM phosphate buffer at pH 7.0. The Hb showed quasi-reversible electrochemical behavior with a formal potential of -50 mV (vs. SCE),cathodic and anodic peaks were not observed using the bare carbon paste electrode. This shows that CdO-NPs acts as a facilitator of electron transfer from the redox species of Hb to the electrode surface and vice versa. These results are in line with the previous work that explains the behavior of nanoparticles as the facilitators of electron transfer [6].

Figure 4: Cyclic voltammograms, using (a) the CdO NPs carbon paste electrode in 50mM phosphate buffer and (b) Hb/ CdO -NPs/ carbon paste electrode in 50mM phosphate buffer (scan rate: 100mVs–1).
To further investigate the Hb characteristics at the Hb/CdO-NPs/graphite electrode,the effect of scan rates on the Hb voltammetric behavior was studied in detail. The baseline subtraction procedure,for the cyclic voltammograms,was obtained in accordance with the method reported by Bard and Faulkner (2001). A linear dependence of anodic and cathodic peak currents on the scan rates is shown in Figures 5A and B. It can be seen that the redox peak currents increased linearly with the scan rate,the correlation coefficients were 0.995 and high value of slopes were obtained. This phenomenon suggested that the redox process was an adsorption-controlled one and the immobilized Hb was stable and was highly efficient.

Figure 5: (A) Typical cyclic voltammograms of Hb/CdO-NPs/ carbon paste electrode at different scan rates. The voltammograms (from inner to outer) designate scan rates of 100, 200, 300, 400, 500, 600, 700, 800, 900 and 1000 mV s-1, respectively. (B) Dependence of the cathodic (a) and anodic (b) peak currents on the scan rates. All the data were obtained at pH 7.0 and in 50mM phosphate buffer solution
Also no decrease in the peak current was observed after repeated cycles of this experiment. These findings indicate that Hb is strongly adsorbed onto the surface of modified electrode. As could be seen,in the range from 600 to 1000 mV s-1,the cathodic peak potential (Epc) changed linearly versus ln ν with a linear regression equation of y = 0.070x - 0.166 ,r = 0.995. According to the following equation [9]:
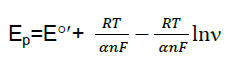
Where α is the cathodic electron transfer coefficient,n the number of electrons,R,T and F are gas,temperature and Faraday constant,respectively (R = 8.314 J mol−1K−1,F = 96485 C/mol,T = 298 K),and αn is calculated to be 0.40. Given 0.3 < α <0.7 in general [10],it could be concluded that n = 1 and α = 0.40. From the width of the peak at mid height and low scan rate,we can also obtain n = 1 [10]. Therefore,the redox reaction between Hb and modified carbon paste electrode is a single electron transfer process. In order to calculate the value of apparent heterogeneous electron transfer rate constant (ks),the following equation was used [9]:
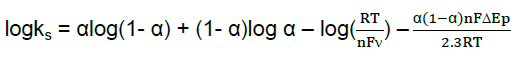
According to Figure 6 ,in a range from 600 to 1000mV/s,the anodic peak potential (Epa) is also linear to ln ν with a linear regression equation of y = 0.077x + 0.198,r =0.993. Ks was calculated to be 1.47 s−1. Figure 7 shows the formal potential of Hb,immobilized onto the CdO-NPs/ carbon paste electrode; in PBS has a strong dependence on the pH of solution. All the changes in the peak potentials and currents with solution pH were reversible in the pH range from 5 to 11. An increase in the solution pH caused a negative shift in both cathodic and anodic peak potentials. Plot of the formal potential versus pH (from 5 to 11) showed a line with the slope of -51 mV pH-1,which was close to the expected value of -57.8 mV pH-1 for a reversible proton-coupled single electron transfer at 291.15 K,indicating that one proton participated in the electron transfer process [11].

Figure 6: Relationship between the peak potential (Ep) and the natural logarithm of scan rate (ln ν) for Hb/CdO-NPs/carbon paste electrode in 50mM PBS (pH 7.0).

Figure 7: Effect of pH on the formal potential of Hb/CdO-NPs/carbon paste electrode.
The cyclic voltammograms of the Hb/CdO-NPs/ carbon paste electrode,in PBS,at pH 7.0,containing different concentrations of NO are shown in Figure 8 . This Hb/CdO-NPs/ carbon paste electrode can facilitate the reduction of oxygen. CVs obtained in an airsaturated solution demonstrate typical electrocatalysis features,suggesting the catalytic reduction of oxygen by the immobilized Hb on the surface of electrode. Compared to CV curves in the absence of oxygen,we have observed increase in the intensity of cathodic peaks. This electrocatalysis follows this reaction mechanism [12,13]:

Figure 8: (A) Cyclic voltammograms obtained at an Hb/CdO-NPs/carbon paste electrode in 50mM phosphate buffer solution (pH 7.0) for different concentrations of NO (from outer to inner, 0, 5×10-8, 5×10-7 and 5×10-6 M, respectively) and (B) magnification of cathodic peak at the range of 0 to -140 mV for the clarity of comprehending is shown.
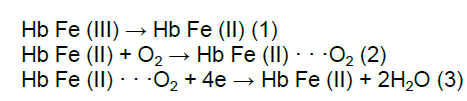
Where Hb Fe(III) and Hb Fe(II) indicate met-Hb and ferrous Hb,respectively,and HbFe(II)···O2 denotes the oxygenated ferrous Hb. NO has high affinity to the heme iron of Hb. Both biochemical and electrochemical studies have revealed that the entry of NO into the distal pocket of Hb disrupts the coordination bond between ferrous heme and O2,which finally produces met-Hb [14]. Therefore,it is anticipated that traces of NO in solution may compete with O2 and modulate the behavior of oxygen’s catalytic reduction. Consistent with this assumption,upon addition of NO to the air saturated solution,the peak shifts to the negative,accompanied by a small attenuation of the peak current (Fig. 8). The attenuation of the peak current obviously comes from the competition between NO and O2. That is,NO repels O2 from the heme site,which decreases the local concentration of O2. The mechanism for the evident peak shift in the presence of NO is more complicated. This might be related to several synergetic processes: the binding of O2 to the heme iron and subsequent catalytic reduction of O2; the competition between O2 and NO; the binding of NO to the iron.
Although the mechanism for this potential shift is still unclear,we expect that this peak shift might be useful for NO measurement. As is expected,successive addition of aliquots of NO leads to the continuous negative peak shifts. Importantly,the peak potential shift is linearly proportional the logarithm of the NO concentration (Fig. 9). A fairly wide linear range is observed to be within 5.0 × 10−8 and 5.0 × 10−6 mol/L. The detection limit,defined from three times the signal-to-noise ratio (S/N = 3),is estimated to be 50nM. Scan rates may affect peak potentials. In this study,a scan rate of 100mV/s was always employed in order to obtain reproducible data. We also note that electrocatalytic efficiency is lowered with the increase of scan rates while lower scan rates lead to longer detection time. This choice of the scan rate of 100mV/s thus seems to be a good compromise although we did not try to optimize it. Oxygen concentration might be another factor that influences peak potentials. In this report,we performed experiments at 25°C and 1atm pressure. Oxygen concentration is known to be 8.3mg/L in this condition [15]. We did not try to alter oxygen concentrations in this preliminary report,however it should be mentioned that oxygen fluctuate may occur in vivo detections. Further investigations should be performed before this NO sensor can be used in real application.

Figure 9: Design of biosensor for determination of NO by the relationship between cathodic peak current of Hb and different concentrations of NO, (scan rate: 100mVs-1).
Acknowledgements
The financial supports of Payam-e-Noor University,isfahan,Iran and Payam-e-Noor University,Yazd for the present project are gratefully acknowledged.
References
- Sanz VC,Mena ML,Gonzalez-Cortes A,Yanez-Sedeño P,Pingarron JM (2005). Development of a tyrosinase biosensor based on gold nanoparticles-modified glassy carbon electrodes: Application to the measurement of a bioelectrochemical polyphenols index in wines. J. M. Anal. Chim. Acta. 528,1-8.
- Campuzano S,Serra B,Pedrero M,Villena F,Pingarron JM (2003). Amperometric flow- injection determination of phenolic compounds at self-assembled monolayer-based tyrosinase biosensors. J. Anal. Chim. Acta. 494: 187-197.
- Salimi A,Sharifi E,Noorbakhsh A,Soltanian S (2006). Direct voltammetry and electrocatalytic properties of hemoglobin immobilized on a glassy carbon electrode modified with nickel oxide nanoparticles. Electrochem. Commun.,8: 1499–1508
- Giovanelli D,Lawrence NS,Wilkins SJ,Jiang L,Jones TGJ,Compton RG (2003). Anodic stripping voltammetry of sulphide at a nickel film: towards the development of a reagentless sensor. Talanta,61: 211- 220.
- Turner AFP (2007). Biochemistry - Biosensors sense and sensitivity. Am. Assoc. Advance. Sci.,290,1315-1317.
- Rezaei-Zarchi S,Saboury AA,Norouzi P,Hong J,Ahmadian S,Ganjali MR,Moosavi-Movahedi AA,Moghaddam AB,Javed A (2007). Use of silver nanoparticles as an electron transfer facilitator in electrochemical ligand-binding of haemoglobin. J. Appl. Electrochem.,37: 1021–1026.
- Ch. Fan,X. Liu,J. Pang,G. Li,H. Scheer. (2004),Highly sensitive voltammetric biosensor for nitric oxide based on its high affinity with hemoglobin,Analytica Chimica Acta 523,225–228.
- Dempsey E,Diamond D,Collier A (2004). Development of a biosensor for endocrine disrupting compounds based on tyrosinase entrapped within a poly(thionine) film. Biosens. Bioelectron.,20: 367-377.
- Laviron E (1979). General expression of the linear potential sweep voltammogram in the case of diffusionless electrochemical systems. J. Electroanal. Chem. 101,19-28.
- Ma HY,Hu NF,Rusling JF (2000). Electroactive Myoglobin Films Grown Layer-by-Layer with Poly(styrenesulfonate) on Pyrolytic Graphite Electrodes. Langmuir J. Electroanal. Chem.,16: 4969-4975.
- Shang L,Liu X,Zhong J,Fan C,Suzuki I,Li G (2003). Fabrication of ultrathin,protein-containing films by layer-by-layer assembly and electrochemical characterization of hemoglobin entrapped in the film. Chem. Lett.,32,296-297.
- J.N. Younathan,K.S. Wood,T.J. Meyer,Inorg. Chem. 31 (1992) 3280.
- F.A. Cotton,G. Wilkinson,Advanced Inorganic Chemistry,5th ed.,Wiley,New York,1988.
- R.F. Eich,T. Li,D.D. Lemon,D.H. Doherty,S.R. Curry,J.F. Aitken,A.J. Mathews,K.A. Johnson,R.D. Smith,G.N. Phillips Jr.,J.S. Olson,Biochemistry 35 (1996) 6976.
- T. Haruyama,S. Shiino,Y. Yanagida,E. Kobatake,M. Aizawa,Biosens. Bioelectron. 13 (1998) 763.


Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences