Introns in Phylum Nemertea
James J. Youngblom
Dept. of Biological Sciences California State University, Stanislaus One University Circle Turlock, CA 95382 USA
Abstract
Ribbon worms are members of a taxonomic group (phylum Nemertea) for which little information regarding gene structure is available. This study looked at intron number, intron length, intron position, and intron/exon junctions for the introns of eight genes from the milky ribbon worm, Cerebratulus lacteus. A total of 22 introns were present in the eight genes, averaging approximately 1200 base pairs in length. All intron/exon junctions contained consensus splice site sequences. Intron placement in these eight nemertean genes was compared to introns in homologous counterparts of other species. There were numerous cases where an intron from C. lacteus shared the same position and phase with introns from homologous genes in various other animals, including vertebrates. There was one case where an intron from C. lacteus shared the same position and phase with introns from homologous genes in an assortment of organisms, including vertebrates,other invertebrates, and higher plants.
Research highlights: Eight genes in the nemertean C. lacteus contain an average of 2.75 introns Eight genes in C. lacteus have introns with an average length of 1200 base pairs Twenty-two introns in C. lacteus all contain consensus splice sites Nemertean intron position and phase are often found to be conserved across kingdoms
Keywords
Cerebratulus, intron evolution, splice junctions, intron placement, Nemertea.
1. Introduction
Nemerteans, commonly called ribbon worms, are soft-bodied, unsegmented, vermiform invertebrates found worldwide [1, 2]. All are members of the same taxonomic group, phylum Nemertea [3]. They are protostome animals (lophotrochozoans) [4]. Most ribbon worms are marine animals; a few species are located in fresh water or found on moist tropical land [5]. Ribbon worms are mainly carnivores or scavengers. All ribbon worms have a unique feature called a proboscis that can be quickly shot out of an anterior opening near the mouth.The proboscis coils around and immobilizes prey. Nemerteans range in length from less than one centimeter to greater than thirty meters [6]. The body of nemerteans is highly extensible and can extend many times its normal length [3]. They are a small animal group with around 1270 species named and described [7]. New species continue to be described on a regular basis [8, 9]. This paper describes deciphering genes of Cerebratulus lacteus- the milky ribbon worm. The nucleic acids analyzed in this paper were isolated from an organism collected off the coast of New England.
Introns are noncoding sequences located within eukaryotic genes [10, 11,12,13].Introns are spliced out after gene transcription but before the mRNA leaves the nucleus for translation. Exons are the segments that remain after splicing. Exons and introns alternate in the premature mRNA (pre-mRNA). The spliceosome is the multi-subunit RNA/protein complex that is required for the removal of introns from most eukaryotic pre-mRNAs [14]. Spliceosomal introns are the dominant type of eukaryotic intron and absent from prokaryotic organisms but present in all major eukaryotic lineages [15, 16]. Introns numbers range from an average of ~ nine introns per gene in humans to less than one intron per gene in many single-celled eukaryotes [17].Certain properties of introns are undoubtedly liabilities- they require excess DNA and RNA synthesis and enhance the eukaryotic mutation rate in a number of different ways. On the other side, introns may provide a mechanism for evolution of new proteins and increase the number of protein isoforms generated by a given gene through the use of alternative splicing [18, 19].
This paper looks specifically at introns in eight C. lacteus genes- the first significant look at intron structure and number in the phylum Nemertea. A recent search of Genbank nucleotides turned up information on only 4 introns from 2 nemertean genes [20]. In this paper eight nemertean genes were analyzed including 22 new introns. Intron placement in these nemertean genes was compared to introns in homologous counterparts. When available, the homologous counterparts included vertebrate and invertebrate animals, higher plants, and a fungus.
2. Materials and Methods
2.1 cDNA cloning and Analysis
Live specimens of C. lacteus were obtained though the Aquatic Resources Division of the MBL (Marine Biological Laboratory, Woods Hole, MA). A whole nemertean worm cDNA library was constructed by Amplicon Express® (Pullman, WA). C. lacteus cDNAs were directionally cloned into the EcoRI and XhoI sites of the plasmid vector pBluescript II SK(+) and propagated in E. coli. strain DH10B. The library titer was 9.0 x 105 cfu/ml. The library was amplified and is stored at –80 C. Pure colonies were picked, grown, and cDNA containing plasmids isolated using Qiagen’s QIAprep ® Spin Miniprep kit. The sizes of the plasmid cDNA inserts were determined by restriction enzyme digests of the plasmid or by PCR amplification of the multiple cloning site followed by agarose gel electrophoresis. Plasmid cDNAs inserts > 0.6 kb were chosen for further analysis. Purified plasmids and primers were sent to the sequencing center at San Diego State University (Microchemical Core Facility, San Diego, CA) or to the University of Nevada (Nevada Genomics Center, Reno, NV) for sequencing reactions and gel electrophoresis. The first sequencing pass on each strand was primed using pBluescript plasmid sequences adjacent to the cloning site. Additional sequencing reactions were primed with internal primers developed using the primer design tool Primer 3 [21]. All cDNAs were sequenced end to end, both strands. Sequencing files were analyzed using the freeware package Chromas Lite (https://www.technelysium.com.au/ chromas_lite.html) or Finch TV (https://www.geospiza.com). DNA files were stored, analyzed, compared, and manipulated using the Biology Workbench (https://workbench.sdsc.edu), provided by the San Diego Supercomputer Center.
2.2 Genomic DNA Isolation and Analysis
C. lacteus genomic DNA was isolated from one adult individual. The specimen was sacrificed by immersion in 95% ethanol. Approximately 100 mg of lateral midbody was removed with a clean razor blade. The sample was frozen in liquid nitrogen and pulverized with mortar and pestle until the material resembled medium sized sand grains. A total of 35 mg of the pulverized tissue was utilized for the DNA extraction. The DNA was extracted using the Tissue DNA Kit (E.Z.N.A. protocol for Tissue) purchased from Omega Bio-Tek, Inc., and 50 micrograms of C. lacteus high molecular weight genomic DNA was recovered.
Selected cDNAs were chosen for intron analysis. Intron organization was investigated by designing 4-8 staggered, overlapping primer pairs based on the cDNA sequence. The primer pairs were utilized to PCR amplify the cDNA fragment and a fragment of C. lacteus genomic DNA. For each pair of primers, the PCR amplicons from the cDNA and genomic DNA were compared via 2% agarose gel electrophoresis (50 ml mini-gels). Genomic DNA fragments were purified (QIAquick PCR Purification Kit, Qiagen, Inc.®) and submitted to the Nevada Genomics Center, Reno, NV for sequencing. The Biology Workbench (workbench.sdsc.edu) and the interactive web tools from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov) were particularly useful for finding homologous protein and genomic DNA sequences. A protein multiple sequence alignment (ClustalW) was used to identify homologous amino acids.
3. Results and Discussion
3.1 cDNA Identification
Eight cDNAs from the nemertean Cerebratulus lacteus were deciphered. Each cDNA included a long open reading frame. The number of amino acids ranged from 169 to 329 amino acids. The cDNA was identified by using the predicted peptide sequence to search Swiss-Prot for homologous proteins (Table 1).
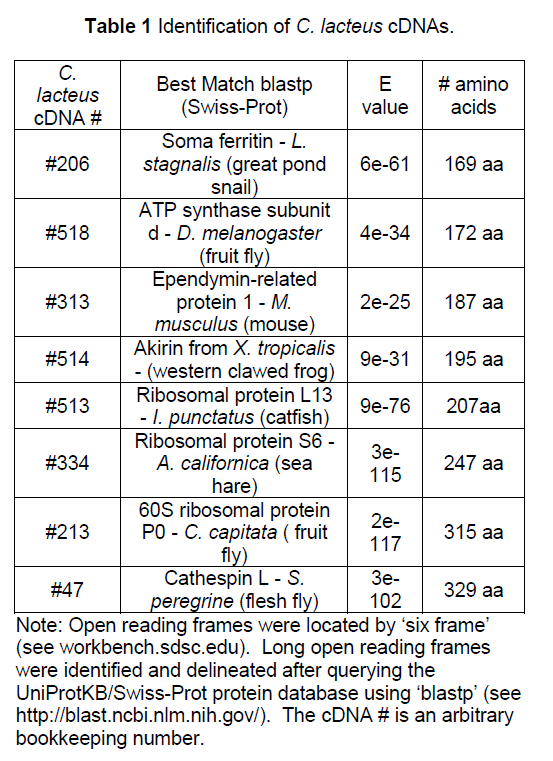
3.2 Intron Analysis
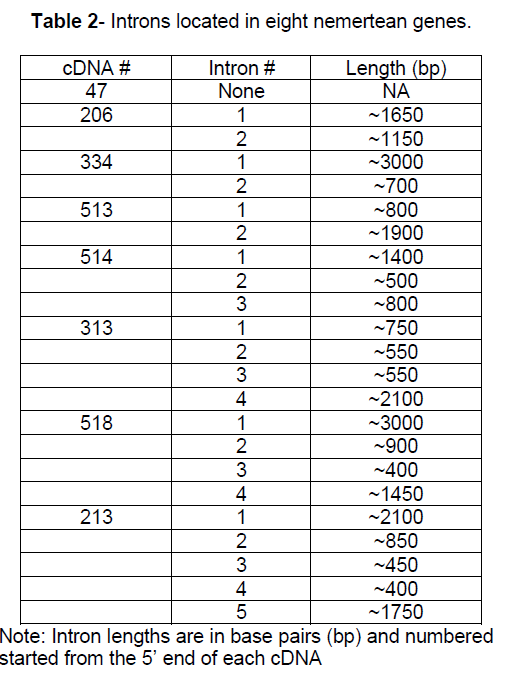
Using the cDNA sequence data, PCR primers were designed for genomic DNA amplification. Introns were identified by comparing genomic DNA amplicons to cDNA amplicons. The genomic DNA fragments were partially deciphered, including each intron/exon junction to identify introns (Table 2). Intron size ranged from ~400 bp to ~ 3000 bp. The average intron number in these eight genes was 2.75 introns/gene. The number of introns identified per gene ranged from 0 to 5. Twenty- two introns were identified with an average length of ~ 1230 bp.
The intron/exon splice junctions identified here conform to the typical eukaryotic splice sites. All introns sequences had a 5’ GT splice donor and a 3’ AG splice acceptor (Figure 1). Other nucleotides around the splice site showed strong conservation.

Figure 1:Consensus sequences for the splice junctions of 26 nemertean introns. Shown in the top section are the consensus for the last nucleotide of the exons and the first 6 nucleotides of the introns. Shown in the lower section are the consensus for the last 6 nucleotides of the introns and the first nucleotide of the exons. The bold lines represent the exon/intron junctions. Twenty-two of the intron junctions were determined by the author and four by Vandergon et al.
An investigation of intron positions was initiated for these 8 nemertean genes. The position of the introns in the C. lacteus genes was compared to the position of the introns in homologous genes. A relevant comparison requires identification of homologous genes. In addition when comparing intron positions in homologous genes, it is critical that homologous amino acid codons can be identified. DNA and protein sequences were available that allowed intron positions to be contrasted between the four of the eight nemertean genes and introns positions in the homologous genes for 2 vertebrates species, 2 invertebrate species, 2 higher plant species, and a fungal species. For the other 4 nemertean genes there were vertebrate and invertebrate homologous sequences available but no counterparts available for higher plants and/or fungus (Figure 2).

Figure 2:Triangles represent intron positions in 8 nemertean genes and homologous counterparts. The intron is located immediately after the amino acid indicated in the column heading. Whole numbers represent phase 0 introns. Phase 1 and phase 2 introns are represented by xxx.1 or xxx.2, respectively. U represents the 5’ untranslated region. All numbers are relative to the amino acid position in the C. lacteus version of the protein. Common names of the organisms are as follows: H. sapiens = human, X. tropicalis = western clawed frog, D. melanogaster = fruit fly, C. elegans = nematode roundworm, C. lacteus = milky ribbon worm, A. thaliana = mouse-ear cress (flowering plant), O. sativa = rice, S. purpuratus = sea urchin, D. rerio = zebra fish, S. kowalevskii = acorn worm, M. brevicollis = choanoflagellate, N. crassa = bread mold, E. intestinalis = fungal parasite (microsporidia). ? = information unavailable.
Since the discovery of introns in the late 1970s, biologists have theorized on the origin of spliced genes[22, 23, 24, 25]. It is agreed that there is a strong but imperfect correlation between intron size and intron number with eukaryotic complexity- generally the more complex the organism, the longer and more numerous the introns [26]. When asked to explain this correlation, there is much disagreement. Two dichotomous theories dominated the initial debates: introns-early [27] and introns-late [28].Proponents of introns-early (also called the exon theory of genes) argue that the earliest genes (before the evolution of eukaryotic organisms) had introns and that they played a pivotal role in gene evolution [29, 30, 31]. In this view early genes evolved into more complex genes as small exons were recombined into larger multiexon units. In introns-early, bacteria have no introns today as they have slimmed down their genomes and lost their introns over the eons. The debate shifted subtly with the discovery that there are different types of introns [32, 33, 34, 35]. Bacteria have the simpler self-splicing introns but lack spliceosomal introns. Debates regarding intron evolution are predominantly revolving around the evolution of spliceosomal introns and the spliceosome [36]. To some, the lack of a single spliceosomal intron in any prokaryotic genome despite thousands deciphered indicates that bacteria never had complex introns- hence the emergence of a contrasting theory, introns-late. In introns late, bacteria never had spliceosomal introns[37, 38]. In this view, spliceosomal introns evolved along with eukaryotic complexity. Most proponents of introns-late argue that introns played no positive role in eukaryotic evolution but are parasitic segments of DNA that progressively invaded eukaryotic genomes. In this view the predominant evolutionary activity is not intron loss, but intron gain. Proponents of introns-late have declared that as additional genes have been deciphered and more introns and intron placements discovered, intron evolution without intron gain seems unlikely. With only intron losses to account for the observed introns, the number of interruptions in the ancestral gene needs to be unreasonably high.
As more and more complete genomes are deciphered, the intron debate is shifting. There is general agreement that introns were present in the earliest eukaryotic (or pre-eukaryotic) organisms. The burgeoning field of comparative genomics has resulted in numerous publications hypothesizing intron loss and intron gain as factors determining current eukaryotic intron placement and number [39, 40]. The predicted rates vary considerable across eukaryotic taxa [41]. There is particularly strong evidence to support recent intron gains in various organisms and gene families.
4. Conclusions
Despite progress in uncovering the dynamic nature of introns, there is still much to explore. The structure of ancestral genes is unknown. Hence one can only use analysis of modern genes to make inference regarding the evolution of eukaryotic genes. The structure of introns in the phylum Nemertea is unknown. This paper presents a first detailed analysis of introns in phylum Nemertea. The introns discovered here ranged in size from ~400 to ~3000 bp for the twenty-two introns present in eight genes (average 2.75 introns per gene). Neither the intron size or intron number is unusual for an invertebrate organism [42,43] The analysis of introns from genes with deciphered homologous counterparts permitted cross kingdom analysis of intron placement. As revealed here, the structure of introns in the nemertean C. lacteus conforms to the same intron structure found in most other eukaryotic genes, suggesting that these introns undergo canonical splicing. The placement of introns in genes of C. lacteus reveal homologous intron placement (same amino acid, same phase) across various taxonomic kingdoms. How can this be explained? Most intron biologists take this, and other similar analyses, as evidence for spliceosomal introns in the common ancestor of eukaryotic organisms. Coupled with the modern-day evidence for recent intron losses and gains, neither of the domineering hypotheses (introns-early or introns-late) is likely to be substantiated. If introns were present in the last common ancestor of prokaryotic and eukaryotic cells and have been jumping into and out of eukaryotic lineages, introns-continual is the more accurate description.
Acknowledgements
This work was funded by RSCA, California State University, Stanislaus (USA) and BRC of the Dept. of Biological Sciences, California State University (USA). Numerous undergraduates research assistants participated in the generating of the data, particularly Alyssa Cifelli, Ahmad Eltejaye, Stephen Jacobs, Austen Jelcick, Peter Lindbeck, Nelson Membreno, Denisse Monroy, Jennifer Morotti, Nicole Reeves, Craig Speilman, Brian Stubblefield, and Sarah Tait.
References
- Gibson, R. (1995). Nemertean species and genera of the world: an annotated check-list of original names and description citations, synonyms, current taxonomic status, habitats and recorded zoogeographic distribution. J. Nat. Hist. 29, 271–562.
- Turbeville, J.M. (2002). Progress in Nemertean Biology: Development and Phylogeny. Integr. Comp. Biol. 42, 692-703.
- Thollesson, M., Norenburg, J.L. (2003). Ribbon worm relationships: a phylogeny of the phylum Nemertea. Proc. Biol. Sci. 270, 407-415.
- Turbeville, J.M. (1986). An ultrastructural analysis of coelomogenesis in the Hoplonemertine Prosorhochmus americanus and the Polychaete Magelona sp. J. Morphol. 187, 51–60.
- Brusca, R.C., Brusca, G.J. (2003). Invertebrates (3rd ed.) Sinauer Associates, Inc. Sunderland, Mass., USA.
- Turbeville, J.M. (1996). Nemertini, Schnurwürmer. In Spezielle Zoologie—Teil 1: Einzeller und Wirbellose Tiere (Westheide W., Rieger R.M., editors). Gustav Fischer Verlag, Stuttgart (Germany). pp. 265-275.
- Kajihara H., Chernychev A.V., Sichun S., Sundberg P., Crandall F. (2008). Checklist of nemertean generea and species (Nemertea) published between 1995 and 2007. Spec. Div. 13: 245-274.
- Costello, M.J., Wilson, S.P. (2011). Predicting the number of known and unknown species in European seas using rates of description. Glob. Ecol. and Biogeogr. 20, 319-330.
- Sundberg, P., Strand, M. (2010). Nemertean taxonomy - time to change lane? J. Zool. Syst. Evol. Res. 48: 283-284.
- Berget, S.M., Moore, C., Sharp, P.A. (1977). Spliced segments at the 5' terminus of adenovirus 2 late mRNA. Proc. Natl. Acad. Sci. USA 74, 3171–3175.
- Chow, L.T., Gelinas, R.E., Broker, T.R., Roberts, R.J. (1977). An amazing sequence arrangement at the 5’ ends of adenovirus 2 messenger RNA. Cell 12, 1–8.
- Evans, R.M., Fraser, N., Ziff, E., Weber, J., Wilson, M., Darnell, J.E. (1977). The initiation sites for RNA transcription in Ad2 DNA. Cell 12, 733-739.
- Goldberg, S., Schwartz, H., Darnell, J. E. (1977). Proc. Natl. Acad. Sci. USA 74, 4520-4523.
- Nilsen, T. (2003). The spliceosome: the most complex macromolecular machine in the cell?. Bioessays 25, 1147–9.
- Collins, L., Penny, D. (2005). Complex Spliceosomal Organization Ancestral to Extant Eukaryotes. Mol. Biol. Evol. 2, 1053-1066.
- Nixon, J.E.J. , Wang, A., Morrison, H.G. , McArthur, A.G., Sogin, M.L., Loftus, B.J., Samuelson, J. (2002). Proc. Natl. Acad. Sci. USA 99, 3701-3705.
- Logsdon, J.M. Jr, Stoltzfus, A., Doolittle, W.F. (1998b). Molecular evolution: recent cases of spliceosomal intron gain? Curr. Biol. 8, R560-R563.
- Black, D.L. (2003). Mechanisms of alternative pre-messenger RNA splicing. Annu. Rev. Biochem. 72, 291–336.
- Gilbert, W. (1985). Genes-in-pieces revisited. Science 228, 823-4.
- Vandergon, T.L., Riggs, C.K., Gorr, T.A., Colacino, J.M. and Riggs, A.F. (1998). The mini-hemoglobins in neural and body wall tissue of the nemertean worm, Cerebratulus lacteus. J. Biol. Chem. 273, 16998-17011.
- Rozen,S., Skaletsky,H. (2000). Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132, 365–386.
- Jeffares, D.C., Mourier, T. , Penny, D. (2006). The biology of intron gain and loss. Trends Genet. 22, 16-22.
- Lynch, M. (2002). Intron evolution as a population-genetic process. Proc. Natl. Acad. Sci. USA 99, 6118-6123
- Roy, S.W., Gilbert, W. (2006). The evolution of spliceosomal introns: patterns, puzzles and progress. Nature Rev. Genet. 7, 211-221.
- Simpson, A.G. B., MacQuarrie, E.K., Roger, A.J. (2002). Eukaryotic evolution: Early origin of canonical introns. Nature 419, 270.
- Lynch, M., Conery, J.S. (2003). The Origins of Genome Complexity. Science 302, 1401-1404.
- Gilbert, W., Glynias, M. (1993). On the ancient nature of introns. Gene 135, 137-144.
- Logsdon, J.M., Jr. (1998a). The recent origins of spliceosomal introns revisited. Curr. Opin. Genet. Dev. 8, 637–648.
- Darnell, J.E. Jr. (1978). Implications of RNA-RNA splicing in evolution of eukaryotic cells. Science 202, 1257–1260.
- Doolittle, W.F. (1978). Genes in pieces: were they ever together? Nature 272, 581–582.
- Gilbert, W. (1978). Why genes in pieces? Nature 271: 501.
- Brody, E., Abelson, J. (1985). The "spliceosome": yeast pre-messenger RNA associates with a 40S complex in a splicing dependent reaction. Science 228, 963-967.
- Cech, T.R. (1990). Self-splicing of group I introns. Annu. Rev. Biochem. 59, 543–68.
- Frendewey, D., Keller, W. (1985). Stepwise assembly of a pre-mRNA splicing complex requires U-snRNPs and specific intron sequences. Cell 42, 355-367.
- Grabowski, P. J., Seiler, S. R., Sharp, P.A. (1985). A multicomponent complex is involved in the splicing of messenger RNA precursors. Cell 42, 345-353.
- Ahmadinejad, N., Dagan, T., Gruenheit, N., Martin, W., Gabaldón, T. (2010). Evolution of spliceosomal introns following endosymbiotic gene transfer. BMC Evol. Biol. 10, 57-67.
- Palmer, J.D., Logsdon, J.M. Jr. (1991). The recent origins of introns. Curr. Opin. Genet. Dev. 1, 470–477.
- Cavalier-Smith, T. (1991). Intron phylogeny: a new hypothesis. Trends Genet. 7, 145–148.
- Rogozin, I.B., Wolf, Y.I., Sorokin, A.V., Mirkin, B.G., Koonin, E.V. (2003). Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr. Biol. 13, 1512-1517.
- Stajich, J.E., Dietrich, F.S., Roy, S.W. (2007). Comparative genomic analysis of fungal genomes reveals intron-rich ancestors. Genome Biol. 8, R223.
- Roy, S.W., Gilbert, W. (2005). Rates of intron loss and gain: implications for early eukaryotic evolution. Proc. Natl. Acad. Sci. USA 102, 5773-5778.
- Colbourne, J.K., Pfrender, M.E., Gilbert, D., Thomas, W.K., Tucker, A., Oakley, T.H., Tokishita, S., Aerts, A., Arnold, G.J., Basu, M.K., Bauer, D.J., Cáceres, C.E., Carmel, L., Casola, C., Choi, J., Detter, J.C., Dong, Q., Dusheyko, S., Eads, B.D., Fröhlich, T., Geiler-Samerotte, K.A., Gerlach, D., Hatcher, P., Jogdeo, S., Krijgsveld, J., Kriventseva, E.V., Kültz, D., Laforsch, C., Lindquist, E., Lopez, J., Manak, J.R., Muller, J., Pangilinan, J., Patwardhan, R.P., Pitluck, S., Pritham, E.J., Rechtsteiner, A., Rho, M., Rogozin, I.B., Sakarya, O., Salamov, A., Schaack, S., Shapiro, Shiga,H., Y., Skalitzky, C., Smith, Z., Souvorov, A., Sung, W., Tang, Z., Tsuchiya, D., Tu, H., Vos, H., Wang, M., Wolf, Y.I., Yamagata, H., Yamada, T., Ye, Y., Shaw, J.R., Andrews, J., Crease, T.J., Tang, H., Lucas, S.M., Robertson, H.R., Bork,P., Koonin, E.V., Zdobnov, E.V., Grigoriev, I.V., Lynch, M., Boore, J.L. (2011). The Ecoresponsive Genome of Daphnia pulex. Science 331, 555-561.
- Koonin, E. (2009). Intron-Dominated Genomes of Early Ancestors of Eukaryotes. J. Hered. 100, 618-623.


Open Access Journals
- Aquaculture & Veterinary Science
- Chemistry & Chemical Sciences
- Clinical Sciences
- Engineering
- General Science
- Genetics & Molecular Biology
- Health Care & Nursing
- Immunology & Microbiology
- Materials Science
- Mathematics & Physics
- Medical Sciences
- Neurology & Psychiatry
- Oncology & Cancer Science
- Pharmaceutical Sciences